Vibrational and Electronic Energy Levels of Polyatomic Transient Molecules
Marilyn E. Jacox
Summary
A critical evaluation and summary of experimental vibrational and electronic energy level data for neutral and ionic transient molecules and high temperature species possessing from three to sixteen atoms is presented. Although the emphasis is on species with lifetimes too short for study using conventional sampling techniques, there has been selective extension of the compilation to include data for isolated molecules of inorganic species such as the heavy-metal oxides, which are important in a wide variety of industrial chemical systems. Observations in the gas phase, in molecular beams, and in rare-gas and diatomic molecule matrices are evaluated. The types of measurement surveyed include conventional and laser-based absorption and emission techniques, laser absorption with mass analysis, and photoelectron spectroscopy.
Outline
- Introduction
- Scope of Database
- Types of Measurement
- Guide to the Compilation
- Abbreviations
- Acknowledgment
- General References
- Figures
Introduction
Most chemical processes--including not only laboratory and industrial chemical syntheses but also those which occur in flames, propellant systems, the initiation of energetic materials, atmospheric pollution, chemical vapor deposition, and plasma processing--consist of a complicated sequence of interrelated reactions in which neutral and charged molecular fragments play essential roles. Although these fragments are present in only very small concentration, they are highly chemically reactive. If a specific molecular fragment is removed from the system, as by introducing a scavenger molecule, the reactions in which that fragment participates stop. Other parts of the overall process continue, resulting in very significant changes in product distribution and yield.
In the early studies of complex chemical processes, it was necessary to postulate mechanisms involving such transient intermediates, present in concentrations too small for direct detection. Conventional end product analysis aids in the selection of suitable mechanisms, but generally does not yield a complete description of the system. Consequently, the improvement of industrial chemical processes often is achieved by semiempirical experimentation. The determination of the detailed chemical mechanism would, in turn, permit the development of rational strategies for removing undesired products and enhancing the yield of the desired species.
In recent years, there has been great progress in the development of techniques suitable for monitoring chemical reaction intermediates. Molecular spectroscopy is especially well suited to this task. Optical detection can be used not only for gas-phase measurements, but also for studies of processes that occur on surfaces or in the condensed phase. It also permits remote sensing, an important advantage. A wide variety of recently developed laser-based spectroscopic detection schemes are not only highly sensitive but also space and time specific. Although the development of spectroscopy-based diagnostics for chemical reaction systems is in its infancy, already the laboratory application of sophisticated sampling and observation techniques has yielded a wealth of vibrational and electronic spectral data for reaction intermediates.
Earlier Data Compilations
For many years, the most important source of vibrational and electronic energy level data for small polyatomic reaction intermediates was the compilation of spectroscopic data for small polyatomic molecules (3-12 atoms) given by Herzberg [1]. To meet the need for an updated, critically evaluated compilation, a series of publications [2,3,4] have appeared in the Journal of Physical and Chemical Reference Data, culminating in the publication in 1994 of a monograph [5] (Vibrational and Electronic Energy Levels of Polyatomic Transient Molecules) which presented evaluated spectral data for more than 1550 small polyatomic transient molecules, defined as species that have a lifetime of less than a few minutes in the pressure range (typically 0.1 to 1.0 Torr) encountered in their production. Vibrational fundamentals in the ground and excited electronic states and radiative lifetimes were included. To aid in spectral identification, the principal rotational constants were also given to three decimal places. These tables provide the basis for this database, designed to supplement the published compilation by providing a capability for rapid searches by molecule or wavenumber.
Recent Progress
The rapid growth in the scientific literature concerned with the spectroscopic study of transient molecules and with their detection in chemical reaction systems continues. Since the October 1993 cutoff in the data evaluation for the monograph, substantive new spectroscopic data have been published for many species included in it, and the first data have become available for almost 2000 other molecules. There has been especially great progress in the spectroscopic characterization of transient species produced by the reaction of metal atoms with oxygen and other small molecules. Despite this rapid progress, many gaps remain in our knowledge of the energy levels of the species represented, and many new and potentially important transient molecules are still to be discovered. This compilation attempts to provide a comprehensive, critically evaluated summary of vibrational and electronic energy level data for small polyatomic transient molecules, in order to support further research and new technologies such as those of plasma processing and chemical vapor deposition.
Scope of Database
A critical evaluation and summary of experimental vibrational and electronic energy level data for neutral and ionic transient molecules and high temperature species possessing from three to sixteen atoms is presented. By early 2003, evaluated data were available for approximately 3500 molecules, and the published version encompassed non only the original monograph [5], but also two supplements [6,7]. The Chemistry WebBook database brings these results together in an ongoing effort to provide evaluated data to the scientific community. Although radiative lifetimes and principal rotational constants are not presently included in this version of the database, references to the original literature concerned with then are given.
Stable Molecules and High-Temperature Species
Data have been selectively included for some molecules which are important in environmental and industrial chemical reaction systems but which can be studied only with difficulty using conventional sampling techniques because of the ease with which they decompose, rearrange, or polymerize. Also included are data derived from spectra of many high-temperature species, such as metal oxides, studied in molecular beams and in rare-gas matrices. Unfortunately, with these few exceptions it is not possible to include data for stable molecules. However, the spectra of many of these species are relatively well established, and sources of data such as the tables of Herzberg [1] remain extremely useful. In obtaining spectral identifications with the help of the present data, it is crucial that the possible contribution of the absorptions or emissions by a stable molecule also be considered.
Types of Measurement
Ground-State Vibrational Spectra
Gas-Phase Measurements
Studies in the gas phase offer the potential for the most precise, detailed measurements. Because of the high chemical reactivity of transient molecules, it is difficult to obtain gas-phase survey infrared spectra of them. The well known advantages of Fourier transform infrared measurements, coupled with sophisticated digital data handling procedures, have permitted the acquisition of gas-phase survey spectra for a number of transient molecules.
Vibrational Frequencies from Rydberg Transitions
Although vibrational frequencies of ground-state molecular ions have frequently been estimated from structure in Rydberg transitions of the parent neutral species, in this compilation vibrational frequencies of the ions are not inferred from these Rydberg transitions. Many of these transitions have residual valence character, resulting in significant variations in vibrational frequencies from one Rydberg state to another.
Matrix Isolation Measurements
As in the earlier compilations, spectral data obtained for molecules trapped in dilute solid solution, with the solvent a rare gas or a small covalent molecule, are included. The application of this sampling technique, known as matrix isolation, for the stabilization and spectroscopic study of uncharged reaction intermediates has recently been reviewed [8]. Because nitrogen and the rare gases are transparent through the entire infrared spectral region, matrix isolation measurements provide a potentially valuable survey tool. In these matrices, infrared absorptions are typically sharp, with half band widths between 0.1 and l cm-1. Rotational structure is, with few exceptions, quenched. Multiple trapping sites occur, often resulting in the appearance of several absorption maxima--usually one or two of which predominate--over a range of a few cm-1.
Matrix Shifts for Uncharged Covalently Bonded Molecules
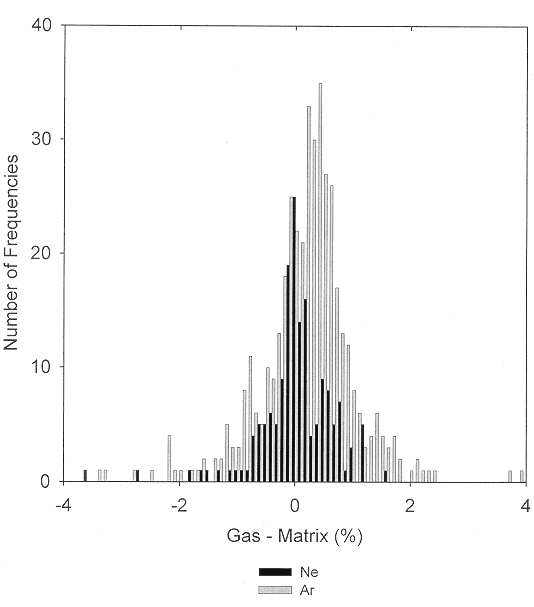
Matrix shifts for covalently bonded molecules trapped in solid neon or argon often are quite small. A comparison [9] of the positions of the ground-state vibrational fundamentals of over two hundred diatomic molecules observed in the gas phase and in nitrogen and rare-gas matrices has shown that, typically, the smallest matrix shift occurs for neon matrix observations, with successively greater matrix shifts for the heavier rare gases and for nitrogen. Except for very weakly bonded molecules and for the alkali metal and Group IIIa halides, matrix shifts of most diatomic molecules isolated in solid argon are smaller than 2%. Similar conclusions resulted from a comparison of neon- and argon-matrix shifts for the ground-state vibrational fundamentals of larger molecules [10]. The generalization that matrix interactions are minimal for neon and that they increase as the mass of the rare gas is increased and become even more important for nitrogen and most other small molecule matrices is supported both by experimental observations on larger molecules and by ab initio calculations [11] for the weakly bonded CaH2 and CaF2 molecules complexed with the rare gases and with nitrogen. Figure 1 compares the observed matrix shifts for the ground-state fundamental vibrations of transient molecules trapped in solid neon and argon. For neon matrices, the maximum in the distribution lies near 0.0%, and for argon matrices, near 0.2%. For both neon and argon matrices, fewer than one-tenth of the matrix shifts are greater than 1%.
Data are beginning to appear for molecules trapped in a hydrogen matrix. Insufficient information is available for generalization on the magnitude of matrix shifts in this medium. For the few species heretofore studied, including several transient molecules present in this compilation, the matrix shifts have been comparable to those in a neon matrix.
Many other matrix materials have also been employed for spectroscopic studies. However, complications due to reaction or to relatively strong interaction (e.g., hydrogen bonding) of the transient molecule with the matrix frequently occur. Therefore, observations in such media as solid hydrocarbons and aqueous solutions and studies of condensed reaction products without an inert carrier have been excluded.
Matrix Shifts for Molecular Ions
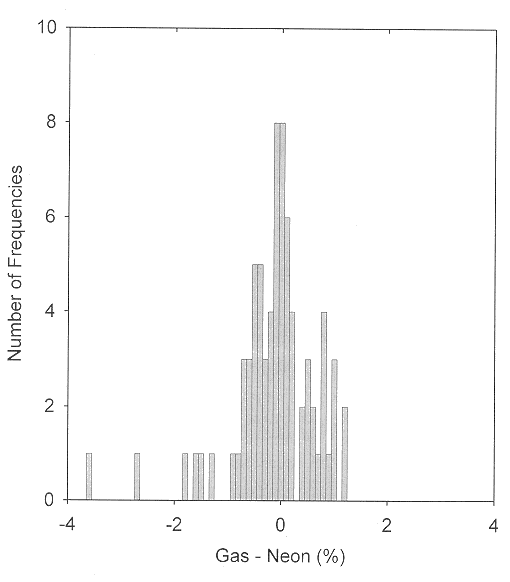
For molecular ions, neon is the matrix of choice. Polarization and charge-transfer interactions become successively more important for molecules isolated in the heavier rare gases. Charge delocalization sometimes also occurs for ionic species trapped in the rare gases [12,13,14]. The anomalously large matrix shift for ν3 of ClHCl- may be attributed to this phenomenon. As is shown in Figure 2, data for small cation species trapped in solid neon are consistent with the matrix shift generalizations given above. Only twelve comparisons are available for molecular cations observed both in the gas phase and trapped in solid argon. The absolute values of five of the observed matrix shifts are greater than 1%. For several other vibrations of molecular ions which have been observed in both neon and argon matrices but not in the gas phase, there are deviations greater than 1% between the neon- and argon-matrix frequencies. Very few comparisons are possible for molecular anions. A number of these species have been generated in rare-gas (usually argon) matrices by charge transfer between a precursor molecule and an alkali metal atom. Recent studies of such species as CO2- and SO2- generated instead by photoionization and/or Penning ionization and trapped in solid neon indicate that shifts on the order of 50 cm-1 may be attributed to the relatively strong interaction of the anion with the nearby alkali metal cation. On the other hand, when the uncharged molecule has a relatively large electron affinity, as is true for C2 and for NO2, charge transfer occurs at a relatively great separation, and a substantial fraction of the anion population may be trapped in sites in which interaction with the alkali metal cation is minimal.
Matrix Shifts for Ionic Bonds
Matrix shifts for vibrations associated with ionic bonds are often considerably larger than those associated with uncharged molecules or with intramolecular vibrations of molecular ions. Criteria for inclusion of data for species that include an ionic bond are exemplified by the selection process for the heavy-metal oxides. Often the stable dioxide structures include an M+O2- species with significant covalent bond character for the attachment of M+, evidenced by a substantial shift in the O2- stretching fundamental as M+ is varied. Such species are included in the compilation. On the other hand, there is little evidence for substantial metal-atom participation in the vibrations characteristic of the O3- moiety of M+O3-. Accordingly, spectral data are given for O3-, but not for M+O3-.
Electronic Spectra
Gas-Phase Electronic Spectra
Gas-phase studies of the electronic spectra of transient molecules were for many years much more readily conducted than were studies of ground-state vibrational spectra. The concentration of transient molecules in flames, chemiluminescent reactions, or various types of discharge may be sufficiently high for spectroscopic study. The photographic plate provides a cumulative detector for visible and ultraviolet radiation. Much of the electronic spectral data summarized in these tables was obtained using conventional gas-phase ultraviolet absorption or emission spectroscopy, which affords the potential for both a broad spectral survey and very high resolution.
Flash Photolysis
Flash photolysis permits the observation of relatively high concentrations of transient species at short time intervals after the flash. Because the products are generally formed with much less internal energy than is typical of systems with detectable emission spectra, the absorption spectra obtained in flash photolysis studies are more readily analyzed. Furthermore, the time-resolved detection used in flash photolysis studies provides information on the rates of formation and disappearance of transient molecules in the system.
Laser-Based Techniques
More recently, a wide variety of laser-based techniques have also been used for electronic spectral observations, often with exceptionally high detection sensitivity. In recent years, very high resolutions have been achieved in laser-based spectroscopic measurements. Since a given laser is tunable over a relatively limited spectral region, laser studies of transient molecules are greatly aided by the availability of survey spectra obtained using other techniques. The spatial configuration of the laser beam makes it an extremely powerful tool for studies of the energy levels of molecules in molecular beams. If the molecule of interest is present in a supersonic molecular beam, excited rotational and vibrational energy levels can be very effectively depopulated, and the absorption spectrum of the molecule is greatly simplified. The spatial configuration of the laser beam also makes it amenable to the development of probes for chemical reaction intermediates not only in the laboratory but also in the environment and in industrial processes.
Laser studies may be broadly classified according to whether the interaction of the molecule with the laser beam(s) is followed by photon or mass detection. Photon-based observations are amenable to remote sensing applications. Because pulsed lasers offer an exceptionally wide range of time specificity, they are very useful for determining radiative lifetimes and rates of elementary chemical reactions. The coupling of lasers with mass detection can lead to the identification of transient molecules for which survey spectra are not available. Among the mass-based detection schemes are photofragment spectroscopy and resonance-enhanced multiphoton ionization (REMPI).
REMPI provides a powerful tool for mapping the Rydberg transitions of transient molecules. Whereas laser-excited fluorescence measurements depend upon the presence of electronic energy levels which decay by photon emission, all molecules possess Rydberg energy levels. REMPI measurements depend on multiphoton excitation into a suitable electronic energy level, most often one of Rydberg character. The selection rules may permit excitation of levels which are not accessible by one-photon excitation from the ground state. The range of tunability of the laser is multiplied by the number of photons required for the excitation of the Rydberg level, significantly broadening the spectral regin which can be probed with a given laser. When the parent molecule is a free radical, almost all of the mass signal is generally found to arise from the parent cation, with very little fragmentation.
Photoelectron Spectroscopy
Much valuable information on the energy levels of molecular cations has been obtained from photoelectron spectroscopy. These tables include selective coverage of the voluminous literature on photoelectron spectroscopic measurements. The number of stable molecules that possess more than six atoms for which photoelectron spectra have been reported is too great to permit the inclusion of low-to-moderate resolution photoelectron spectral data for molecular cations with more than six atoms. Those who need such data for larger molecules may find the reviews by Turner et al. [15], Rabalais [16], and Kimura et al. [17] helpful. Several criteria are important in determining whether a given reference should be included in the present work. The first of these is resolution. In the few instances in which high resolution photoelectron data are available, these data are heavily weighted. Where direct spectroscopic observation is possible, the measurements generally are of considerably higher precision than are the photoelectron data, which are then omitted from the tables. A second criterion is the availability of adiabatic ionization potentials. In order to obtain information on the positions of electronic transitions from photoelectron spectral data, it is necessary to subtract the first ionization potential from the energy of the photoelectron band. Where there is little change in the molecular geometry in the transition, the difference between the vertical ionization potentials gives a reasonable approximation to the position of the electronic transition. However, this is not the general case. Therefore, priority is given to papers that include adiabatic ionization potentials.
For most photoelectron spectroscopic transitions, structure has not been resolved. Many of these states are dissociative. Further information on the dissociation products can be obtained from values of the appearance potentials for various products in photoionization studies on the parent molecule. Such studies are beyond the scope of this review. The tables of published ionization and appearance potentials by Lias and co-workers [18,19] and the on-line version included in the Chemistry WebBook constitute a valuable source of information on the appearance potentials of photofragments.
Matrix Shifts in Electronic Spectra
The range of tunability of visible and ultraviolet lasers, like that of infrared lasers, is limited. Therefore, a preliminary survey using conventional gas-phase and/or matrix-isolation spectroscopic studies is often desirable. A comparison of the positions of the electronic band origins of diatomic molecules in the gas phase and in rare-gas and nitrogen matrices has been published [20]. As in the determination of ground-state vibrational energy levels, neon is the matrix material of choice, with a sharp maximum at 0.0% in the distribution of matrix deviations for valence transitions of covalently bonded molecules. In argon-matrix observations, most such band origins are shifted by less than 2% from the gas-phase values. At the somewhat higher temperatures often used for electronic spectral observations in matrices of the heavier rare gases or of nitrogen, relatively broad phonon bands become prominent. The blue shift of the phonon maximum from the zero-phonon line in absorption measurements, and the red shift in emission measurements, typically amount to approximately 1 to 1.5%. Rydberg transitions of molecules in matrices often are greatly broadened and experience much larger shifts. Further details of the behavior of electronic transitions of matrix-isolated molecules have previously been discussed [3,8,20].
Guide to the Compilation
Considerable effort has been expended to provide a critical evaluation of the data. However, for many species the available data are meager. The identities of some species have been proposed on the basis of chemical evidence. While such evidence may be quite compelling, it is not definitive. Many examples could be cited in which a spectrum was later reassigned to characteristic impurities in the sample. Where chemical evidence has provided a reasonable basis for the assignment of vibrational or electronic bands to a transient molecule, data have been included in this compilation, in the hope that further testing of the assignment will be facilitated.
While every effort has been made to make these tables as complete as possible, for various reasons omissions do occur. There remains some selectivity in the coverage of electronic spectral data for larger molecules. Where low-resolution photoelectron spectral data have been superseded by spectroscopic observations with appreciably higher resolution and greater precision, often the photoelectron data have not been cited. Candidate molecules or energy levels may also have been inadvertently omitted. Suggestions of additions or needed revisions to the data to be included in subsequent extensions of this database are welcome, as are inquiries regarding new data added after the cutoff date for this compilation.
Molecular Formulas
Molecular formulas are used in this compilation. In order to permit a compact index, an attempt has been made to provide as much structural information as possible in a minimal amount of space. This restriction is especially severe for larger molecules. The following formula abbreviations have been used:
| br | bridged |
| cyc | cyclic. If parentheses follow, only the atoms enclosed in them are included in the ring. |
| c | cis |
| t | trans |
Isotopic Species
Where heavy isotopic peaks are resolved, data are given for the most abundant isotopic species (e.g., 7Li, 11B, 35Cl, 79Br). Data are included for both the normal and the fully deuterium-substituted molecule, except that data are also given for the partially deuterium-substituted species of H3+ and H3.
Units
Except where otherwise indicated, the units of all quantities in these tables are cm-1.
Error Estimates
Error estimates are those of the authors of the original literature. When the uncertainty is not explicitly indicated, the value is given to the estimated number of significant figures. Where vibrational frequencies have been determined with a precision greater than two decimal places, the tabulated values have been rounded off.
Excited Electronic States
Symmetry
The heading for each electronic state gives its symmetry, the point group to which the molecule belongs in that electronic state, and, where available, references to the determination of a quantitative molecular structure. For C2v molecules, there is potential ambiguity in the definition of the molecular symmetry axes. The convention in which the x axis is chosen perpendicular to the plane of the molecule, recommended by the Joint Commission for Spectroscopy of IAU and IUPAP [21], has been adopted. Often this has required the interchange of published assignments of energy levels with B1 and B2 symmetry.
Assignments Based on Photoelectron Spectroscopy
Most authors of papers on photoelectron spectroscopy have proposed assignments for the various photoelectron bands, using arguments based on molecular orbital theory and often on semiempirical or ab initio calculations. These assignments have been included in the present tables. Where several conflicting assignments have been given in the literature, an attempt has been made to choose the most satisfactory one. Generally, the assignments of photoelectron spectra have been made with the presumption that the point group to which the molecular cation belongs is the same in all of its excited states. Structural data for these excited states are extremely rare. Therefore, the molecular point group which has been adopted in the analysis of the photoelectron spectrum is given in these tables. In practice, it is likely that there is some variation in excited-state molecular symmetries. Thus, a bent molecular ion may become linear in some of its excited states. For highly symmetric species, Jahn-Teller distortion may reduce the molecular symmetry.
Energy of the Electronic Transition
The energy of the electronic transition follows the state designation and symmetry information. Where possible, T0, the energy separation between the electronic energy level of interest and the ground electronic, vibrational, and rotational states of the molecule, is given. However, where only low resolution data or photoelectron data are available, often only band maxima have been given in the literature. With photoelectron data, T0 is derived by subtracting the value of the first ionization potential from that of the higher ionization potential which corresponds to the state of interest. When data for the first adiabatic ionization potential are available, the subscript "x" is used to indicate that the first adiabatic ionization potential is known but that the higher ionization potential is measured to the peak maximum; the subscript "xx" implies that the energy difference between the higher and the first absorption maximum was used. If the first photoelectron transition has a gradual onset, a better value of the first ionization potential may have been obtained from photoionization data or from the extrapolation of Rydberg series in the spectrum of the parent molecule. Supplementary sources of data for the first ionization potential are cited in the tables. However, if the difference between the first adiabatic ionization potential obtained in the photoelectron spectrum and that obtained in other measurements amounts to only 10 or 20 meV, the photoelectron spectroscopic value is used, because of the advantage of a consistent set of measurements. Where threshold energies differ by one quantum in a vibrational progression, a best value for the ionization potential is chosen which coincides with the most probable position of the vibrationally unexcited transition. Because of inherent uncertainties in the determination of higher ionization potentials in many photoelectron spectral measurements, photoelectron peaks above about 18 eV are often omitted.
As in the tables of Herzberg [1], T0 values are given to the center of multiplet structure. For doublet states, the two components differ by ±A (the spin-orbit splitting constant), and the energy difference is measured from the average of the two bands, whereas for triplet states the three components fall at 0, ±A with respect to the position from which the band energy is measured. This convention is also followed here unless specific states are given. However, in matrix isolation absorption and laser excitation studies only the lowest component is accessible. Except for transitions with relatively small values of A, this is also likely to be true for studies using cooled molecular beams. Often these latter studies give T0 values for the lowest energy component with a precision better than that to which A is known.
Wavelength Range of the Electronic Transition
The wavelength range (nm) in which various electronic transitions have been observed is also tabulated. This range is a composite of the values typical of absorption and emission observations. Laser-excited fluorescence studies often include both excitation and resolved emission measurements. Since the position of the band origin is given, ambiguity should not arise. For information on the range in which the band system is observed for a given type of measurement, see the original literature cited for that measurement technique.
Ground- and Excited-State Vibrations
Vibration Numbering Conventions
The format of the vibrational tables is similar to that used in the earlier compilations. The vibrational numbering convention is that used by Herzberg [1]. Within a given symmetry species, vibrations are numbered starting with the highest frequency. The same convention is followed for deuterated species. Therefore, a given type of vibration may be numbered differently for the deuterated than for the unsubstituted molecule. For triatomic molecules, the bending vibration is always designated as ν2. For aromatic molecules, an alternate vibrational numbering scheme developed by Wilson [22] has often been used in the literature. Where both the Herzberg and the Wilson numbering schemes have been used for the published data, the Herzberg numbering is adopted, and the Wilson numbering is sometimes shown in parentheses. For a few species, only the Wilson numbering has been used. To avoid confusion, this is retained in the present tables, and the use of the Wilson numbering is specifically indicated.
Where possible, the values of ΔG(1/2), the separation between the v = 0 and v = 1 levels for the vibration of interest, have been used.
Renner-Teller Interaction
If a bending fundamental is split by Renner-Teller interaction, the position of the unperturbed fundamental is given. Where specific components of such a split fundamental have been studied, they may also be listed, with the transition designated in an explanatory note. For a more complete treatment of the Renner effect and definitions of the associated parameters, see the discussion by Herzberg [1] and the references cited for the molecule of interest.
Inversion Splitting
A few of the species in these tables possess out-of-plane vibrations that have resolved inversion splitting structure. For these, the specific component for which the vibrational frequency is reported is designated in an explanatory note.
Intensities
Relative intensities of vibrational bands are dependent on the technique used for the measurement. When possible, the relative intensities of ground-state infrared absorptions are included. It is not feasible to give the corresponding relative intensities for other types of observation. Relative intensity abbreviations include:
| vw | very weak |
| w | weak |
| m | medium |
| s | strong |
| vs | very strong |
| sh | shoulder |
| br | broad |
Abbreviations
Standard Abbreviations for Transition Energies
One or more of five standard abbreviations may be associated with vibrational or electronic transition energies:
| H | (1/2)(2ν) |
| I | Component of an inversion doublet |
| L | Lower bound |
| T | Tentative assignment or approximate value |
| U | Upper bound |
The following abbreviations are used to designate the type of energy separation measured for electronic transitions:
| o | Energy separation between the v = 0 levels of the excited and the ground electronic states |
| x | Energy separation between the band maximum of the excited electronic state and the v = 0 level of the ground state |
| xx | Energy separation between the band maxima of both the excited state and the ground state of a molecular cation, derived from a photoelectron spectrum |
| d | Photodissociation or photodetachment threshold |
| a | Separation of band origin for an excited state of H3 or D3 from the lowest bound state, 2s 2A1´ |
| t | Energy separation between the v = 0 levels of the two excited electronic states involved in the transition. |
Occasionally, when the band origin is too weak to be observed, specific vibrational quantum numbers are given (e.g., 030 for the A state of D2O+).
For ground-state vibrational energy levels of matrix-isolated molecules, the wavenumber column may provide information on species trapped in a site adjacent to the molecule of interest. The method of preparation of a transient molecule may lead to the trapping of, for example, N2 or HF next to the transient molecule, with consequent shifts in the observed vibrational frequencies. Molecular anions have sometimes been stabilized by charge transfer interaction between an alkali metal and a suitable precursor molecule. Usually the vibrational frequencies observed for the anion show a small dependence on the nature of the alkali metal present in the system. Where such interactions are known to be significant, the wavenumber, intensity, and standard abbreviations are followed by a space, then by the formula of the perturbing species. (For the charge transfer systems, the perturber is the alkali metal cation. However, the entry is abbreviated to the atomic symbol for the alkali metal present in the sample.)
Type of Measurement
Many sophisticated laser techniques--frequently employing two or more laser beams--have been used for studies of transient molecules. The laser may be used both in the preparation of the transient molecule and in the detection scheme. For example, ions may be generated by multiphoton ionization and detected by absorption of radiation from a probe laser. Often the developers of such techniques have designated them by complicated acronyms. In these tables, an attempt has been made to avoid relatively lengthy and unfamiliar acronyms by designating only the generic type of detection, using the abbreviations defined below. (Velocity modulation, designated as a separate detection technique in the first of this series of data evaluations [2], is widely used and is considered to be a measurement tool rather than a type of observation. The type of laser used for the absorption measurement in an infrared detection scheme employing velocity modulation is instead specified in these tables.)
| AB | near infrared-visible-ultraviolet absorption |
| CC | color-center laser |
| CR | cavity ringdown |
| DL | diode laser absorption |
| DPI | depletion photoionization |
| DR | double resonance |
| ED | electron diffraction |
| EF | electron-excited fluorescence |
| EM | near infrared-visible-ultraviolet emission |
| ESR | electron spin resonance |
| FD | fluorescence depletion |
| HFD | high frequency deflection |
| IB | ion beam |
| ID | ion drift, ion depletion (see specific reference) |
| IR | infrared absorption (conventional or Fourier transform) |
| LD | laser difference frequency |
| LF | laser-excited fluorescence (excitation and resolved emission) |
| LMR | laser magnetic resonance |
| LS | laser Stark spectroscopy |
| MO | molecular orbital calculations |
| MPD | multiphoton dissociation |
| MPI | multiphoton ionization |
| MW | microwave and millimeter wave |
| ND | neutron diffraction |
| PD | electron photodetachment |
| PE | photoelectron spectroscopy |
| PEFCO | photoelectron-photon coincidence |
| T-PEFCO | threshold photoelectron-photon coincidence |
| PEPICO | photoelectron-photoion coincidence |
| PF | photofragment spectroscopy |
| PI | photoionization |
| PIFCO | photoion-photon coincidence |
| PIR | photoionization resonance |
| PRI | photoinduced Rydberg spectroscopy |
| Ra | Raman |
| SEP | stimulated emission pumping |
| TF | tunable far-infrared laser |
| TPE | threshold photoelectron spectroscopy, including ZEKE detection |
| UV | near infrared-visible-ultraviolet absorption and emission |
Acknowledgment
This work was supported by the Standard Reference Data Program of the National Institute of Standards and Technology.
General References
- G. Herzberg, Molecular Spectra and Molecular Structure. III. Electronic Spectra and Electronic Structure of Polyatomic Molecules (Van Nostrand, Princeton, NJ, 1966).
- M. E. Jacox, J. Phys. Chem. Ref. Data 13, 945 (1984).
- M. E. Jacox, J. Phys. Chem. Ref. Data 17, 269 (1988).
- M. E. Jacox, J. Phys. Chem. Ref. Data 19, 1387 (1990).
- M. E. Jacox, Vibrational and Electronic Energy Levels of Polyatomic Transient Molecules, Monograph 3, J. Phys. Chem. Ref. Data (1994), 461 pages.
- M. E. Jacox, J. Phys. Chem. Ref. Data 27, 115 (1998).
- M. E. Jacox, J. Phys. Chem. Ref. Data 32, 1 (2003).
- M. E. Jacox, "The Stabilization and Spectroscopy of Free Radicals and Reactive Molecules in Inert Matrices," in Chemistry and Physics of Matrix-Isolated Species, L. Andrews and M. Moskovits, Eds., pp. 75-105 (Elsevier Science Publishers, Amsterdam, The Netherlands, 1989).
- M. E. Jacox, J. Mol. Spectrosc. 113, 286 (1985).
- M. E. Jacox, Chem. Phys. 189, 149 (1994).
- M. Kaupp, P. von R. Schleyer, and H. Stoll, J. Phys. Chem. 96, 9801 (1992).
- H. Kunttu, J. Seetula, M. Räsänen, and V. A. Apkarian, J. Chem. Phys. 96, 5630 (1992).
- M. Räsänen, J. Seetula, and H. Kunttu, J. Chem. Phys. 98, 3914 (1993).
- M. E. Jacox, Chem. Soc. Rev. 31, 108 (2002).
- D. W. Turner, C. Baker, A. D. Baker, and C. R. Brundle, Molecular Photoelectron Spectroscopy (Wiley-Interscience, London, 1970).
- J. W. Rabalais, Principles of Ultraviolet Photoelectron Spectroscopy (Wiley, New York, 1977).
- K. Kimura, S. Katsumata, Y. Achiba, T. Yamazaki, and S. Iwata, Handbook of He I Photoelectron Spectra of Fundamental Organic Molecules (Halstead Press, New York, 1981).
- R. D. Levin and S. G. Lias, "Ionization Potential and Appearance Potential Measurements, 1971-1981," Natl. Stand. Ref. Data Ser., Natl. Bur. Stand. (U. S.), 71 (1982).
- S. G. Lias, J. E. Bartmess, J. F. Liebman, J. L. Holmes, R. D. Levin, and W. G. Mallard, Gas-Phase Ion and Neutral Thermochemistry, J. Phys. Chem. Ref. Data 17, Supplement No. 1 (1988).
- M. E. Jacox, J. Mol. Structure 157, 43 (1987).
- J. Chem. Phys. 23, 1997 (1955).
- E. B. Wilson, Jr., Phys. Rev. 45, 706 (1934).
Figures